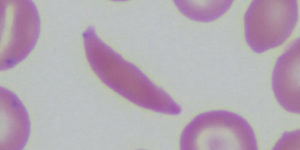
Sickle Cell Disease and Hydroxyurea Treatment
Abstract Introduction: Sickle cell disease (SCD) is a genetic disorder impacting the patient’s haemoglobin. This condition is accompanied by many dangerous phenotypes, which are the result of pathological haemoglobin polymerisation …