
Chronic myeloid leukaemia (CML) is a myeloproliferative disorder caused by BCR-ABL1 igureusion encoding for a tyrosine kinase oncoprotein. Since the introduction of the tyrosine kinase inhibitor (TKI), imatinib, in 2000, CML survival rates have increased, to the point where life expectancy is equal to that of the general population. One obstacle patients face is imatinib resistance. Literature about resistance has mainly focussed on mutations in the Bcr-Abl kinase domain (KD), which have been well described. Areas that have not been as well established include the origin of KD mutations and resistance from mechanisms outside of KD mutations. This review focuses on how KD mutations arise and their mechanisms of resistance and the roles of BCR-ABL1 gene amplification, Erk1, and Lyn kinase in creating resistance outside of the KD. Experimental therapies to combat imatinib resistance are also mentioned. Using database searches to obtain the current literature, this review attempts to determine the current consensus on these topics and highlight areas where research could be beneficial. While the origin of KD-mutations and non-KD resistance is not entirely clear, the many possible causes that have been elucidated thus far have already paved the way for new therapies.
Introduction
Chronic myeloid leukaemia (CML) was the first cancer where the pathological chromosomal abnormality was identified, and is one of the most understood and well-managed cancers [1,2]. CML is a clonal disorder of pluripotent stem cells that results in over-proliferation of mature myeloid cells [3]. Constitutive and aberrant tyrosine kinase activity is responsible for pathological cell proliferation in CML [4].
Before the advent of tyrosine kinase inhibitors (TKIs), 5-year survival rate for patients aged 20-44 was 40%, and less than 20% for patients over 65 years. For patients aged 15-44 diagnosed in 2000, this jumped to 71.6%, increasing to 86.4%, if diagnosed in 2005 [5]. Responsible for these leaps in survival was imatinib mesylate, a TKI approved in 2001 [6]. Imatinib antagonises tyrosine kinase activity by competing with ATP binding to the Bcr-Abl protein, reducing unchecked cell-cycle progression [3]. Imatinib resistance undermines therapy, putting patients at risk, and occurs in approximately 25% of patients [1]. Hence, it is important for doctors and medical students alike to understand that resistance occurs, some of the mechanisms behind resistance and how new pharmacotherapies can combat these. This review summarises the pathophysiology of CML and synthesises the literature around competing theories of imatinib resistance.
Pathophysiology of CML
CML is a myeloproliferative disease caused by a reciprocal translocation between chromosome 9 and 22 (9;22)(q34;q11.2)[1,7]. This creates an abnormal chromosome 22 called the Philadelphia (Ph) chromosome, named after the city it was discovered in in 1960 [2,8]. The oncogenic effects of this translocation are caused by 5’ exons of the BCR (breakpoint cluster region) gene fusing to the 3’ exons of ABL1 (Abelson tyrosine protein kinase 1) [3]. This creates the BCR-ABL1 oncogene on the Ph chromosome encoding for Bcr-Abl tyrosine kinase, or p210BCR/ABL [9-11]. Retroviral insertion of p210BCR-ABL in murine models induces a myeloproliferative disorder similar to CML [11]. However, in less than 10% of cases, gene fusion occurs in different exons creating kinases p190 or p230 depending on where the fusion occurs [12,13]. Bcr-Abl has constitutive tyrosine kinase activity, causing modulated gene transcription, proliferation, and enforced survival of myeloid progenitor cells [14]. Unregulated cells grow and enter the S-Phase of the cell cycle independently of physiological growth factors and avoid apoptosis [8,14,15]. Abl and Bcr-Abl are non-receptor tyrosine kinases that travel between the nucleus and the cytoplasm and phosphorylate proteins via SH2 and SH3 domains [16].
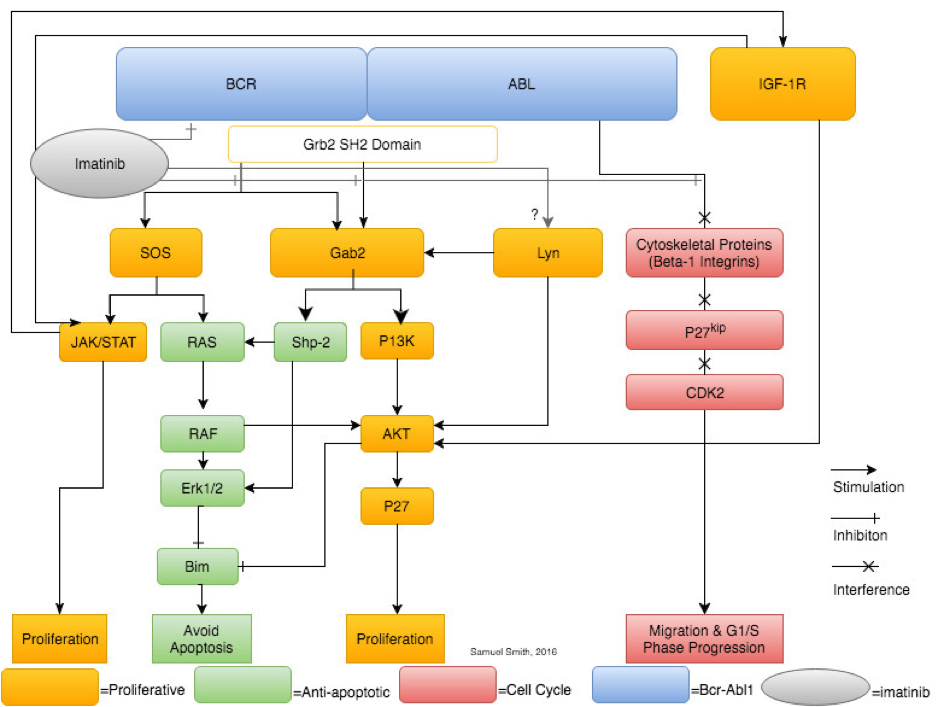
A synthesis and simplification of selected pathways (JAK/STAT, Gab2, Lyn kinase, IGF-1 and β-1 integrin) showing the leukaemogenic downstream effects of Bcr-Abl signalling. Imatinib is shown solely inhibiting Bcr-Abl, however, research shows imatinib therapy also affects Lyn kinase expression and activity.
Imatinib
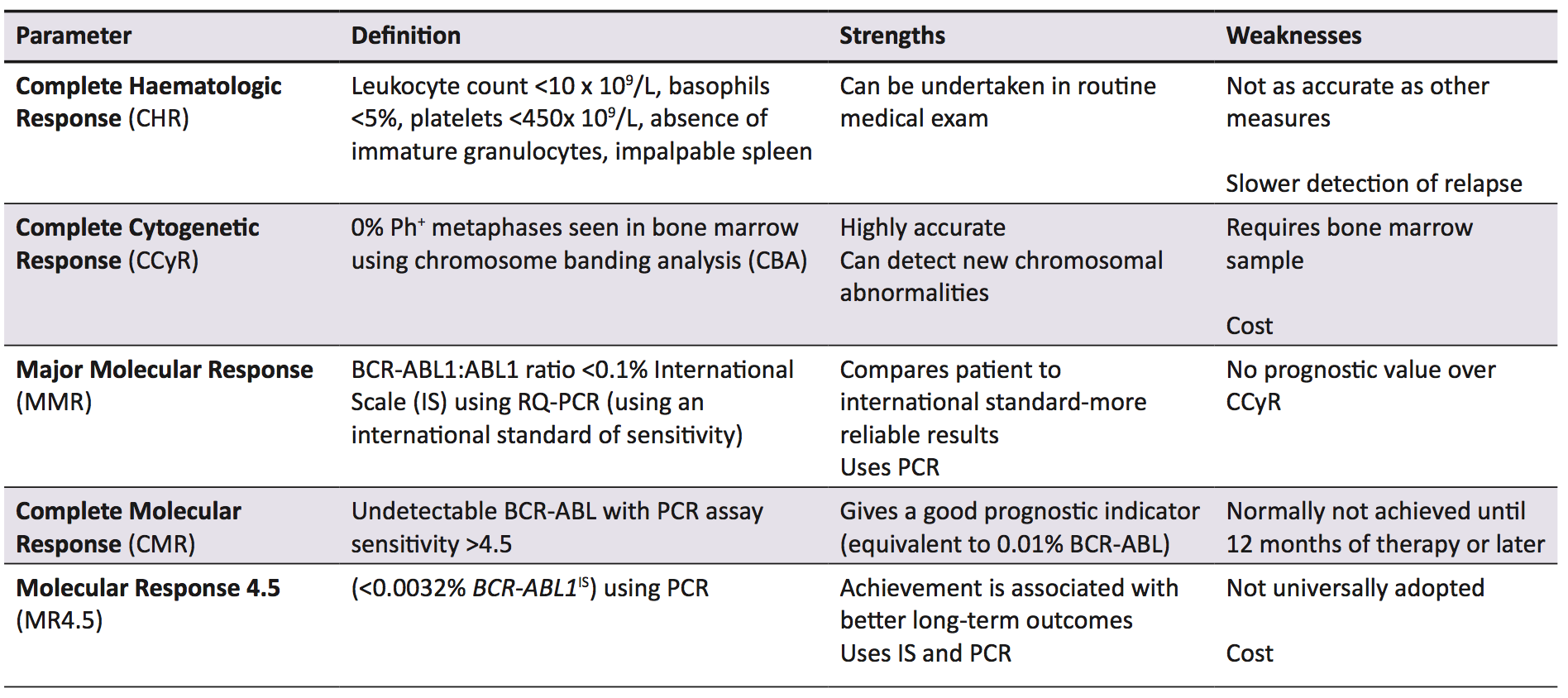
Imatinib is a TKI hailed as a conceptual breakthrough in targeted chemotherapy, and is the first line treatment in CML, while also being used in Ph+ acute lymphocytic leukaemia and some gastric cancers [27]. In early evaluation studies, it was found to specifically inhibit cellular proliferation and tumour formation of Bcr-Abl expressing cells by 92%-98% [28]. Imatinib is effective when compared to other early treatments for CML, increasing the likelihood of survival at ten years by 20% when compared to the closest alternative therapy (Figure 2) [29]. The current parameters to measure successful treatment outcomes are listed with definitions in Table 1. In the International Randomized Study of Interferon and STI571, it was found that complete haematological response (CHR), complete cytogenetic response (CCyR), and major molecular response (MMR) scores were superior in imatinib-treated patients compared with interferon-treated patients [30]. Additionally, in the original trials for imatinib approval, three phase II studies involving 1027 patients showed over 90% of patient leukocyte counts returned to normal, and when combined with interferon, 100% of evaluable patients achieved CHR [31].

This figure from the German CML-Study group shows patient survival probability as a function of time after diagnosis in five consecutive randomised treatment optimisation studies.
Mechanism of action
Bcr-Abl has an activation loop, a P-loop with an ATP binding site (in the kinase domain) and substrate anchoring SH2 and SH3 domains [33,34,35]. Imatinib binds specifically to the ATP-binding sites of Bcr-Abl, c-kit, and platelet-derived growth factor receptors, and inhibits their tyrosine kinase activity by both preventing ATP binding and stabilising the activation loop in an inactive conformation [34,36,37]. When Bcr-Abl tyrosine kinase activity is inhibited, there is no downstream signalling and treatment is successful in 77% of all patients [1] and virtually 100% of patients treated in the early stages of the disease [34]. The European LeukemiaNet (ELN) 2013 guidelines use MMR as the marker to indicate success of TKI therapy [32], however, Thompson et al. [1]. have argued that MMR overestimates the number of patients with treatment failure.
Treatment considerations and mechanisms of Imatinib resistance in CML
Failure of TKI therapy is caused by a number of factors, including inappropriate drug choice, patient non-adherence, and drug resistance. There are a number of opinions as to what constitutes treatment failure, but the definition used by this paper is the one put forward by the ELN and European Society for Medical Oncology. Treatment failure depends on which measurement is used; using haematological parameters (testing peripheral leukocyte counts), treatment failure is defined as no haematological response by three months, or any loss of CHR. Using cytogenetic response, however, treatment failure is no cytogenetic response within six months, no CCyr by 18 months, or any loss of CCyr, as detected by CBA [38]. Finally, a sub-optimal molecular response is defined as no MMR by 18 months or loss of MMR at any point using PCR for BCR-ABL1 transcripts [38]. While this review focuses on imatinib (the gold standard in CML therapy) [30], second generation TKIs such as nilotinib, ponatinib, and dasatinib are now also used, both as first line agents and for use in imatinib resistant patients [1,27]. Which TKI to use and at what dose depends on a number of factors, including imatinib sensitivity and which disease phase the patient is in. CML is staged into chronic phase (CP), accelerated phase (AP), and blast phase (BP) (Table 2) [39]. For instance, a patient in AP who has never had a TKI is still treated with imatinib, but if they have taken imatinib and then progressed to AP, a second generation TKI is used [1]. Using imatinib inappropriately could cause treatment failure, while immediately using a second generation TKI or increasing TKI dosages has been found to limit adherence, as well as side effects (Table 2), especially for patients who require more potent TKIs and higher doses [40].


Other research suggests that BCR-ABL1 has the ability to cause self-mutagenesis. Mutation rate and advanced disease phase were correlated, consistent with mutations being related to exposure time to Bcr-Abl activity [34]. One suggested mechanism is production of reactive oxygen species causing genomic instability, shown in vitro and in murine models, but beyond the original studies, no further research has been undertaken [34,57]. While KD mutations are a highly researched area in CML therapy (over 60 unique point mutations have been identified), there remains an information deficit, for example, the prevalence of mutations in specific populations, or randomised controlled trials for TKI choice following imatinib failure [1,58].
Non Bcr-Abl kinase domain mediated resistance
Recent research adds complexity by suggesting there are a number of mutations and events occurring outside of Bcr-Abl KD that impact drug resistance [43].This includes mutations of Bcr-Abl1 outside of the kinase domain, such as BCR-ABL1 amplification, and causes outside of the Bcr-Abl1 protein altogether, such as mesenchymal cells, drug transporters and bypass molecular pathways. Increased BCR-ABL1 expression via gene amplification is found in most TKI resistant cells, whether the mutations are primary, secondary, KD, or non-KD, implying a link between increased expression and resistance [59]. However, increased expression is not the sole cause of non-KD mediated resistance as studies have shown that increasing imatinib concentration in non-mutated, sensitive cells with induced BCR-ABL1 amplification still reduces Bcr-Abl activity, whereas some resistant cells without KD mutations remain resistant at any dose [60].
Extracellular Signal-Regulated Kinase 2 (Erk2) is a Mitogen Activated Protein Kinase (MAPK) and has been implicated in both primary and secondary resistance (i.e. immediate resistance to therapy and resistance that builds over time)[60,61]. In a study of non-KD mutated resistant cells treated with imatinib, Erk2 was found in the nucleus of resistant cells only, and inhibiting Erk2 caused damage to resistant cells [60]. Mechanisms for how Erk2 could cause primary resistance were then elucidated. To achieve this, mutated Ras (which activates Erk2), was virally transduced into sensitive cells that were cultured and treated with imatinib. Using proliferation assays to determine cell survival, it was discovered activating Erk2 gave previously sensitive cells resistance without any prior exposure to imatinib. Erk2 is a key regulator of the pro-apoptotic molecule Bim and it is proposed interactions between Erk2 and Bcr-Abl over-stimulate Erk2 and reduce CML cell apoptosis [62]. Research in 2016 by Wong et al. [63] extended these results to create a pharmacotherapy inhibiting Erk2, showing areas outside of the KD can cause primary and secondary resistance and can be targeted.
Lyn kinase
Lyn kinase is a non-receptor tyrosine kinase regulated by Bcr-Abl. Imatinib resistant but Bcr-Abl KD-mutation negative cells were found to overexpress Lyn kinase following treatment with imatinib [64]. In cell lines from these patients, while imatinib effectively inhibited Bcr-Abl activity, Lyn kinase phosphorylation continued, allowing proliferation to continue. Interestingly, prior to imatinib therapy, there was no consistent difference in Lyn expression between sensitive and resistant cells, but afterward there were consistent distinctions in their control of phosphorylation. This implies imatinib treatment uncouples Lyn expression from Bcr-Abl, leading to resistance. Lyn overexpression can induce a three to fourfold resistance, equal to some KD mutations, yet the mechanisms of its overexpression and how it worsens CML are not yet known [65]. One theory is that because silencing of Lyn kinase induces apoptosis in CML cells, overexpression causes cell survival, signalled through via Gab2 [66,67]. The fact that this effect is not seen in imatinib-naïve CML patient cells supports the idea that Lyn kinase only causes acquired resistance, leaving the mechanisms behind primary resistance a mystery.
Drug transporters
Alterations in drug transporters are yet another mechanism by which medication resistance can occur and will be mentioned briefly. A drug must both reach the target organ in sufficient amounts and be present at an effective therapeutic concentration for it to exert and effect, and both influx and efflux transporters can interfere with these pharmacokinetics [27]. Radiolabelled imatinib assays have determined that the level of kinase inhibition is dependent on the level of uptake and retention of imatinib achieved [68]. Imatinib enters the circulation from the gastrointestinal tract by a member of the organic cation transporter (OCT) family, OCT-1, thus mutations in OCT-1 are thought to contribute to treatment failure [68]. Conversely, imatinib leaves the cell via the p-glycoprotein multidrug resistance protein-1 (MDR1 or ABCB1) [69]. In other drugs, MDR1 overexpression has been confirmed to cause drug resistance by increasing efflux before a therapeutic concentration can be reached, and this is a relationship currently under investigation in CML.
Non-Bcr-Abl KD resistance is not a well-studied area and much research is yet to be undertaken. Two recent CML mutation reviews by Jabbour et al. and O’Hare et al. only provide a brief mention of non Bcr-Abl mutations causing resistance, even though these mutations cause from 10%-40% of TKI resistance [3,70]. In addition, there was much disagreement among researchers concerning molecular pathways to resistance. Erk2 is part of a super-family of MAPKs, other members such as Erk1, Erk5, and P38MAPK, have been considered in imatinib resistance [71,72]. Aceves-Luquero et al. carried out knock-out studies of MAPKs, which identified only Erk2 as having a resistance-inducing effect [52]. Extremely resistant patients require potent TKIs or stem-cell transplantation, both of which greatly affect quality of life, which could be avoided if mechanisms behind resistance were uncovered and targeted treatment developed1.
Limitations
While p210BCR-ABL accounts for 90% of cases of CML, other Bcr-Abl variants were not examined despite their different treatment responses, limiting the applicability of this review. Furthermore, the diagram in Figure 1 is a simplified representation of the pathways associated with Bcr-Abl, especially in the case of JAK/STAT. Only pathways that have been clearly implicated in CML and imatinib resistance by research literature were included. Systematic database searches were used to carry out this review. Spelling and terminology variations that influence search results, for example, “myeloid” and “myelogenous”, were a limiting factor.
Clinical implications
This article holds a number of clinical implications for all medical students, not just the aspiring oncologist. For instance, the prevailing view in oncology is that mutations that confer imatinib resistance occur in the kinase domain. With the explosion of advances in genome sequencing, it is becoming possible to prospectively genetically screen patients to determine whether resistance will occur. If the current wisdom regarding CML resistance prevails, then mutations outside the kinase domain (that have been reported to cause between 10% – 40% of resistance) could be ignored, potentially putting patients at risk of ineffective treatment which could cost them their lives [2]. By investigating and becoming aware of the role of non-KD mutations, doctors could also give more accurate prognoses to patients with these mutations and begin studies looking at the best treatment for these cases (for example, randomised controlled trials comparing current therapy to higher doses of imatinib, or new pharmacological agents altogether). This review also provides a general overview into CML pathophysiology, imatinib pharmacology and chemotherapy resistance, topics every medical practitioner should be very familiar with.
Conclusion
Imatinib is a TKI that revolutionised leukaemia treatment and increased the length and quality of life of CML patients. While it has been known for many years that primary and secondary resistance to imatinib exist, the mechanisms have not been fully explained. While mutations in the Bcr-Abl KD account for the majority of resistance and are well known, what remains unclear is the origin of these mutations, and how resistance occurs without KD mutations. Stem cell mutations and self-mutagenesis are possible explanations for how KD mutation occurs, and gene amplification, Lyn kinase and Erk2 for resistance occurring outside of the KD. Further research identifying key events in downstream pathways will offer new approaches for overcoming all forms of imatinib resistance.
Acknowledgements
The author would like to thank and acknowledge A/Prof Peter Johnson and A/Prof Bill Warren of James Cook University School of Medicine and Dentistry who provided comments and feedback on this paper. The author would also like to thank Dr. Donna Rigano and Miss Shalisa Maisrikrod for their assistance and editing help.
Conflicts of interest
None declared
References
- Thompson PA, Kantarjian HM, Cortes JE. Diagnosis and treatment of chronic myeloid leukemia in 2015. Mayo Clinic proceedings. 2015;90(10):1440-54.
- Nowell P HD. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497.
- Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Haematol. 2014;89(5):547-56.
- An X, Tiwari AK, Sun Y, Ding PR, Ashby CR, Jr., Chen ZS. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leukemia research. 2010;34(10):1255-68.
- Brunner AM, Campigotto F, Sadrzadeh H, Drapkin BJ, Chen YB, Neuberg DS, et al. Trends in all-cause mortality among patients with chronic myeloid leukemia: a surveillance, epidemiology, and end results database analysis. Cancer. 2013;119(14):2620-9.
- Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8(5):935-42.
- JD R. A new consistent chromosomal abnormality in chromc myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290-3.
- Holtz MS, Forman SJ, Bhatia R. Nonproliferating CML CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia. 2005;19(6):1034-41.
- Rafiei A, Mian AA, Doring C, Metodieva A, Oancea C, Thalheimer FB, et al. The functional interplay between the t(9;22)-associated fusion proteins BCR/ABL and ABL/BCR in Philadelphia chromosome-positive acute lymphatic leukemia. PLoS genetics. 2015;11(4):e1005144.
- McLaughlin J, Chianese E, Witte ON. In vitro transformation of immature hematopoietic cells by the P210 BCR/ABL oncogene product of the Philadelphia chromosome. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(18):6558-62.
- Daley GQ, Van Etten RA, Baltimore D. Induction of Chronic Myelogenous Leukemia in Mice by the P210$^{bcr/abl}$ Gene of the Philadelphia Chromosome. Science. 1990;247(4944):824-30.
- Arana-Trejo RM, Ruiz Sanchez E, Ignacio-Ibarra G, Baez de la Fuente E, Garces O, Gomez Morales E, et al. BCR/ABL p210, p190 and p230 fusion genes in 250 Mexican patients with chronic myeloid leukaemia (CML). Clinical and laboratory haematology. 2002;24(3):145-50.
- Chan LC, Karhi KK, Rayter SI, Heisterkamp N, Eridani S, Powles R, et al. A novel abl protein expressed in Philadelphia chromosome positive acute lymphoblastic leukaemia. Nature. 1987;325(6105):635.
- Hershkovitz-Rokah O, Modai S, Pasmanik-Chor M, Toren A, Shomron N, Raanani P, et al. Restoration of miR-424 suppresses BCR-ABL activity and sensitizes CML cells to imatinib treatment. Cancer letters. 2015;360(2):245-56.
- Jonuleit T, Peschel C, Schwab R, van der Kuip H, Buchdunger E, Fischer T, et al. Bcr-Abl kinase promotes cell cycle entry of primary myeloid CML cells in the absence of growth factors. British journal of haematology. 1998;100(2):295-303.
- Bertacchini J, Ketabchi N, Mediani L, Capitani S, Marmiroli S, Saki N. Inhibition of Ras-mediated signaling pathways in CML stem cells. Cell Oncol. 2015;38(6):407-18.
- Shi P, Chandra J, Sun X, Gergely M, Cortes JE, Garcia-Manero G, et al. Inhibition of IGF-IR tyrosine kinase induces apoptosis and cell cycle arrest in imatinib-resistant chronic myeloid leukaemia cells. Journal of cellular and molecular medicine. 2010;14(6B):1777-92.
- Jiang Y, Zhao RC, Verfaillie CM. Abnormal integrin-mediated regulation of chronic myelogenous leukemia CD34+ cell proliferation: BCR/ABL up-regulates the cyclin-dependent kinase inhibitor, p27Kip, which is relocated to the cell cytoplasm and incapable of regulating cdk2 activity. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(19):10538-43.
- Lakshmikuttyamma A, Pastural E, Takahashi N, Sawada K, Sheridan DP, DeCoteau JF, et al. Bcr-Abl induces autocrine IGF-1 signaling. Oncogene. 2008;27(27):3831-44.
- Xie J, Chen X, Zheng J, Li C, Stacy S, Holzenberger M, et al. IGF-IR determines the fates of BCR/ABL leukemia. Journal of hematology & oncology. 2015;8:3.
- Zha J, Lackner MR. Targeting the insulin-like growth factor receptor-1R pathway for cancer therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(9):2512-7.
- Million RP, Van Etten RA. The Grb2 binding site is required for the induction of chronic myeloid leukemia-like disease in mice by the Bcr/Abl tyrosine kinase. Blood. 2000;96(2):664-70.
- Bhatia R, Verfaillie CM. Inhibition of BCR-ABL expression with antisense oligodeoxynucleotides restores beta1 integrin-mediated adhesion and proliferation inhibition in chronic myelogenous leukemia hematopoietic progenitors. Blood. 1998;91(9):3414-22.
- Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf NA, Podar K, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1(5):479-92.
- Goss VL, Lee KA, Moritz A, Nardone J, Spek EJ, MacNeill J, et al. A common phosphotyrosine signature for the Bcr-Abl kinase. Blood. 2006;107(12):4888-97.
- O’Hare T, Deininger MW, Eide CA, Clackson T, Druker BJ. Targeting the BCR-ABL signaling pathway in therapy-resistant Philadelphia chromosome-positive leukemia. Clin Cancer Res. 2011;17(2):212-21.
- Rang HP RJ, Flower RJ, Henderson G. Anti-Cancer Drugs. Rang & Dale’s Pharmacology. 8th Edition ed. Philadelphia, Pennsylvania: Elsevier; 2015. p. 687-8.
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996;2(5):561-6.
- Hehlmann R. CML—where do we stand in 2015? Annals of Hematology. 2015;94(2):103-5.
- O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. The New England journal of medicine. 2003;348(11):994-1004.
- Habeck M. FDA licences imatinib mesylate for CML. The Lancet Oncology. 2002;3(1):6.
- Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872-84.
- Segaliny AI, Tellez-Gabriel M, Heymann MF, Heymann D. Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. Journal of bone oncology. 2015;4(1):1-12.
- O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110(7):2242-9.
- Ren SY, Xue F, Feng J, Skorski T. Intrinsic regulation of the interactions between the SH3 domain of p85 subunit of phosphatidylinositol-3 kinase and the protein network of BCR/ABL oncogenic tyrosine kinase. Experimental hematology. 2005;33(10):1222-8.
- Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer research. 1996;56(1):100-4.
- Kantarjian HM, Cortes JE, O’Brien S, Giles F, Garcia-Manero G, Faderl S, et al. Imatinib mesylate therapy in newly diagnosed patients with Philadelphia chromosome-positive chronic myelogenous leukemia: high incidence of early complete and major cytogenetic responses. Blood. 2003;101(1):97-100.
- Jabbour E, Cortes JE, Kantarjian HM. Suboptimal response to or failure of Imatinib treatment for chronic myeloid leukemia: what is the optimal strategy? Mayo Clinic proceedings. 2009;84(2):161-9.
- World Health Organization Classification of Tumors of Haematopoietics and Lymphoid Tissues. . In: Swerdlow SH CE, Harris NL, et al, editor. Lyon, France: IARC Press; 2008.
- Branford S, Yeung DT, Prime JA, Choi SY, Bang JH, Park JE, et al. BCR-ABL1 doubling times more reliably assess the dynamics of CML relapse compared with the BCR-ABL1 fold rise: implications for monitoring and management. Blood. 2012;119(18):4264-71.
- Australian public assessment report for Imatinib, Nilotinib and Dastinib. In: Administration TG, editor. Woden, ACT, Australia 2014.
- Song KW, Rifkind J, Al-Beirouti B, Yee K, McCrae J, Messner HA, et al. Subdural hematomas during CML therapy with imatinib mesylate. Leukemia & lymphoma. 2004;45(8):1633-6.
- Wei Y, Hardling M, Olsson B, Hezaveh R, Ricksten A, Stockelberg D, et al. Not all imatinib resistance in CML are BCR-ABL kinase domain mutations. Annals of Hematology. 2006;85(12):841-7.
- le Coutre P TE, Varella-Garcia M, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood 2000;95(2):1758-66.
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876-80.
- Elias MH, Baba AA, Azlan H, Rosline H, Sim GA, Padmini M, et al. BCR-ABL kinase domain mutations, including 2 novel mutations in imatinib resistant Malaysian chronic myeloid leukemia patients-Frequency and clinical outcome. Leukemia research. 2014;38(4):454-9.
- Yun SM, Jung KH, Kim SJ, Fang Z, Son MK, Yan HH, et al. HS-438, a new inhibitor of imatinib-resistant BCR-ABL T315I mutation in chronic myeloid leukemia. Cancer letters. 2014;348(1-2):50-60.
- Wylie A, Schoepfer J, Berellini G, Cai H, Caravatti G, Cotesta S, et al. ABL001, a potent allosteric inhibitor of BCR-ABL, prevents emergence of resistant disease when administered in combination with Nilotinib in an in vivo murine model of chronic myeloid leukemia. Blood. 2014;124(21):398.
- Valent P. Imatinib-resistant chronic myeloid leukemia (CML): current concepts on pathogenesis and new emerging pharmacologic approaches. Biologics. 2007;1(4):433-48.
- Ursan ID, Jiang R, Pickard EM, Lee TA, Ng D, Pickard AS. Emergence of BCR-ABL kinase domain mutations associated with newly diagnosed chronic myeloid leukemia: a meta-analysis of clinical trials of tyrosine kinase inhibitors. J Manag Care Spec Pharm. 2015;21(2):114-22.
- Qin Y, Chen S, Jiang B, Jiang Q, Jiang H, Li J, et al. Characteristics of BCR-ABL kinase domain point mutations in Chinese imatinib-resistant chronic myeloid leukemia patients. Annals of hematology. 2011;90(1):47-52.
- Carella AM, Garuti A, Cirmena G, Catania G, Rocco I, Palermo C, et al. Kinase domain mutations of BCR-ABL identified at diagnosis before imatinib-based therapy are associated with progression in patients with high Sokal risk chronic phase chronic myeloid leukemia. Leukemia & lymphoma. 2010;51(2):275-8.
- Pfeifer H, Wassmann B, Pavlova A, Wunderle L, Oldenburg J, Binckebanck A, et al. Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2007;110(2):727-34.
- Gruber FX, Lundan T, Goll R, Silye A, Mikkola I, Rekvig OP, et al. BCR-ABL isoforms associated with intrinsic or acquired resistance to imatinib: more heterogeneous than just ABL kinase domain point mutations? Medical oncology. 2012;29(1):219-26.
- Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106(6):2128-37.
- Skorski T. BCR/ABL regulates response to DNA damage: the role in resistance to genotoxic treatment and in genomic instability. Oncogene. 2002;21(56):8591-604.
- Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer letters. 2008;270(1):1-9.
- Nicolini FE, Corm S, Le QH, Sorel N, Hayette S, Bories D, et al. Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: a retrospective analysis from the French intergroup of CML (Fi(phi)-LMC GROUP). Leukemia. 2006;20(6):1061-6.
- Karimiani EG, Marriage F, Merritt AJ, Burthem J, Byers RJ, Day PJ. Single-cell analysis of K562 cells: an imatinib-resistant subpopulation is adherent and has upregulated expression of BCR-ABL mRNA and protein. Experimental hematology. 2014;42(3):183-91 e5.
- Aceves-Luquero CI, Agarwal A, Callejas-Valera JL, Arias-Gonzalez L, Esparis-Ogando A, del Peso Ovalle L, et al. ERK2, but not ERK1, mediates acquired and “de novo” resistance to imatinib mesylate: implication for CML therapy. PloS one. 2009;4(7):e6124.
- Hartel N, Klag T, Hanfstein B, Mueller MC, Schenk T, Erben P, et al. Enhanced ABL-inhibitor-induced MAPK-activation in T315I-BCR-ABL-expressing cells: a potential mechanism of altered leukemogenicity. Journal of cancer research and clinical oncology. 2012;138(2):203-12.
- Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(40):14907-12.
- Wong SM, Liu FH, Lee YL, Huang HM. MPT0B169, a New Antitubulin Agent, Inhibits Bcr-Abl Expression and Induces Mitochondrion-Mediated Apoptosis in Nonresistant and Imatinib-Resistant Chronic Myeloid Leukemia Cells. PloS one. 2016;11(1):e0148093.
- Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101(2):690-8.
- Wu J, Meng F, Kong LY, Peng Z, Ying Y, Bornmann WG, et al. Association between imatinib-resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. Journal of the National Cancer Institute. 2008;100(13):926-39.
- Ptasznik A, Nakata Y, Kalota A, Emerson SG, Gewirtz AM. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nature medicine. 2004;10(11):1187-9.
- Wu J, Meng F, Lu H, Kong L, Bornmann W, Peng Z, et al. Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood. 2008;111(7):3821-9.
- White DL, Saunders VA, Dang P, Engler J, Zannettino ACW, Cambareri AC, et al. OCT-1–mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108(2):697.
- Illmer T, Schaich M, Platzbecker U, Freiberg-Richter J, Oelschlägel U, Bonin Mv, et al. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia. 2004;18(3):401-8.
- Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA working party on chronic myeloid leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(24):7374-9.
- Buschbeck M, Hofbauer S, Di Croce L, Keri G, Ullrich A. Abl-kinase-sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO reports. 2005;6(1):63-9.
- Ozaki K, Kosugi M, Baba N, Fujio K, Sakamoto T, Kimura S, et al. Blockade of the ERK or PI3K-Akt signaling pathway enhances the cytotoxicity of histone deacetylase inhibitors in tumor cells resistant to gefitinib or imatinib. Biochemical and biophysical research communications. 2010;391(4):1610-5.